Guard1 is an open-label, multinational phase 1/2 clinical trial designed to evaluate the safety and efficacy of AVR-RD-02, an investigational gene therapy, in individuals with Gaucher disease Type 1 (GD1).
Key Eligibility Criteria
Individuals may be able to participate in this trial if they are male or female, are past puberty and:

Are between the ages of 18 and 50 years at the time of screening; and

Have a confirmed diagnosis of Gaucher disease Type 1 based on genotyping, deficient glucocerebrosidase (GCase) enzyme activity, clinical features consistent with Gaucher disease Type 1; and

Have been on stable enzyme replacement therapy (ERT) for a minimum of 24 months, OR have never received ERT or substrate reduction therapy (SRT) or have not received ERT or SRT within the last 12 months.
These are not all the eligibility requirements for the Guard1 trial. Visit ClinicalTrials.gov (study identifier: NCT04145037) for additional eligibility criteria and information.
Study Locations
The Guard1 trial will enroll patients from around the world.
How to Participate

For Physicians:
Call 1-877-330-5214 if you have patients with Gaucher disease Type 1, or at-risk patient family members, who may be interested and eligible for this clinical trial.
or
Complete this form to have someone contact you.

For Patients:
Complete this interest form or call 1-877-330-5214 to speak with someone who can help you determine if you or a family member meet the eligibility requirements and for trial locations.
and
Talk to your doctor or a stem cell transplant specialist about the potential risks and benefits of participating in an investigational gene therapy trial.
About AVR-RD-02
AVR-RD-02 is an investigational gene therapy being developed for patients with Gaucher disease Type 1. AVR-RD-02 is being investigated in clinical trials and has not yet been approved by the U.S. Food and Drug Administration (FDA) or any other regulatory agency, and its safety and efficacy have not yet been established.
Read more about AVROBIO’s gene therapy technology.
About Gaucher Disease
Gaucher disease is a rare, genetic disorder caused by a defect (mutation) in the GBA gene. The faulty GBA gene results in a deficiency of an essential lysosomal enzyme called GCase, which functions to break down a fatty substance called glucocerebroside (GluCer).1 Lysosomes use enzymes to digest and recycle materials, like large molecules such as GluCer, in cells.2 When the GCase enzyme is deficient or does not function properly, GluCer is not removed from the lysosomes of immune system cells called macrophages. The fatty buildup of GluCer causes the macrophages to be enlarged and this results in the progressive signs and symptoms of Gaucher disease Type 1.
Gaucher Disease Type 1 Symptoms3
- Enlargement of the liver and spleen (hepatosplenomegaly)
- Low number of red blood cells (anemia)
- Easy bruising caused by a decrease in blood platelets (thrombocytopenia)
- Chronic fatigue
- Lung disease
- Bone disease such as reduced bone density and strength (osteopenia/osteoporosis), fractures, bone death (avascular necrosis), and severe bone pain/bone crisis
Gaucher disease Type 1 may lead to an increased risk for Parkinson’s disease, peripheral neuropathy, certain cancers, and osteoporosis.3
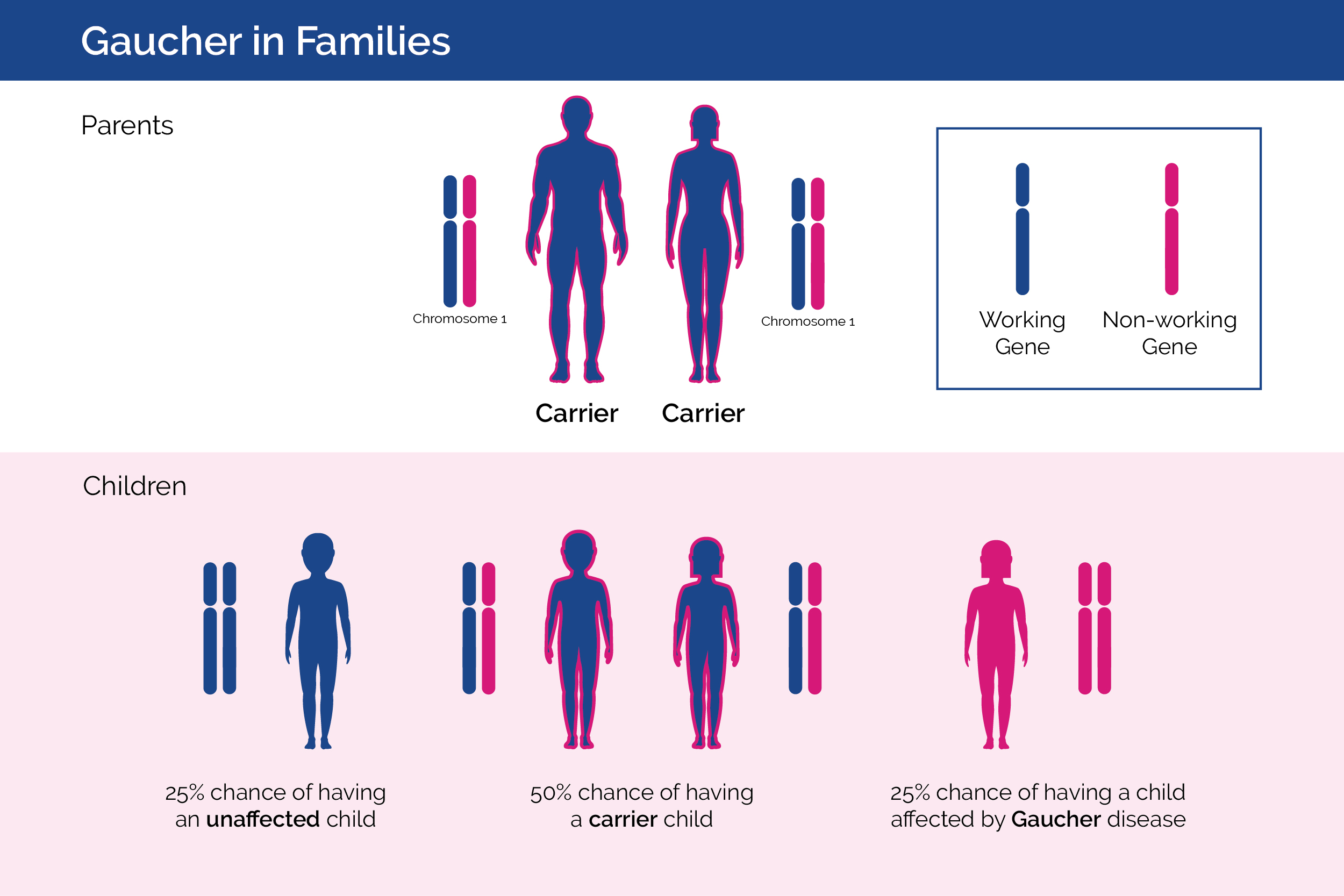
Gaucher in Families
Gaucher disease is inherited, which means that it is passed down from parents to children.4 Gaucher disease is inherited in an autosomal recessive manner. Usually, a person has two working copies of the gene that provides the instruction code for making the GCase enzyme.4 When a person has one working gene and one non-working gene they are called a “carrier” and do not develop Gaucher disease. When two carrier parents have children, each parent will pass one of their genes (either the working gene or the non-working gene) to their child. With each pregnancy, two carrier parents have a 25% chance of having an unaffected child, a 1 in 2 (50%) chance of having a carrier child, and a 1 in 4 chance (25%) of having a child affected by Gaucher disease.5 Males and females are equally affected.
References
1. Futerman AH, Platt FM. The metabolism of glucocerebrosides – From 1965 to the present. Mol Genet Metab. Volume 120, Issues 1-2:22-26 (2017). 2. Shiel WC Jr. Lymphocyte. MedicineNet.com; 2019. Available at: https://www.medicinenet.com/script/main/art.asp?articlekey=4220. Accessed June 20, 2019. 3. Gaucher Disease Type 1. Genetic and Rare Diseases Information Center. https://rarediseases.info.nih.gov/diseases/2441/gaucher-disease-type-1. Accessed October 24, 2019. 4. About Gaucher Disease. Genome.gov. https://www.genome.gov/Genetic-Disorders/Gaucher-Disease. Accessed October 24, 2019. 5. Pastores GM et al. Gaucher Disease. GeneReviews Pagon RA, Adam MP, Ardinger HH, et al., editors. Seattle (WA): University of Washington, Seattle; (2015).